Medical Device Regulations: EAEU vs. Russia
What Is a Medical Device? Defining Medical Products.
Medical devices in Russia and EAEU are classified differently, with the central part of the definition overlapping. The essential difference in the definitions is the drug support for medical devices:
- In Russian legislation, medical devices are those “which function is not implemented by pharmacological, immunological, genetic or metabolic effects on the human body.”
- In EAEU regulations, medical devices are those, which cannot be realized by pharmacological, immunological, genetic or metabolic effects on the human body, however, can be supported by drugs.”
A significant update regarding drug support for medical devices is finally described in the region’s laws, clarifying and harmonizing the current legislation.
What Are the Overlaps and Differences in Medical Device Legislation?
Many discrepancies are noticed in the medical device regulations when comparing them to the national registration procedures. Comparing EAEU and Russian legislation, there are five main differences:
- Territory of circulation
- Authorized Representatives of the Manufacturer responsibility
- Operational documentation language
- Document approval
- Approach to quality, effectiveness, and safety confirmation.
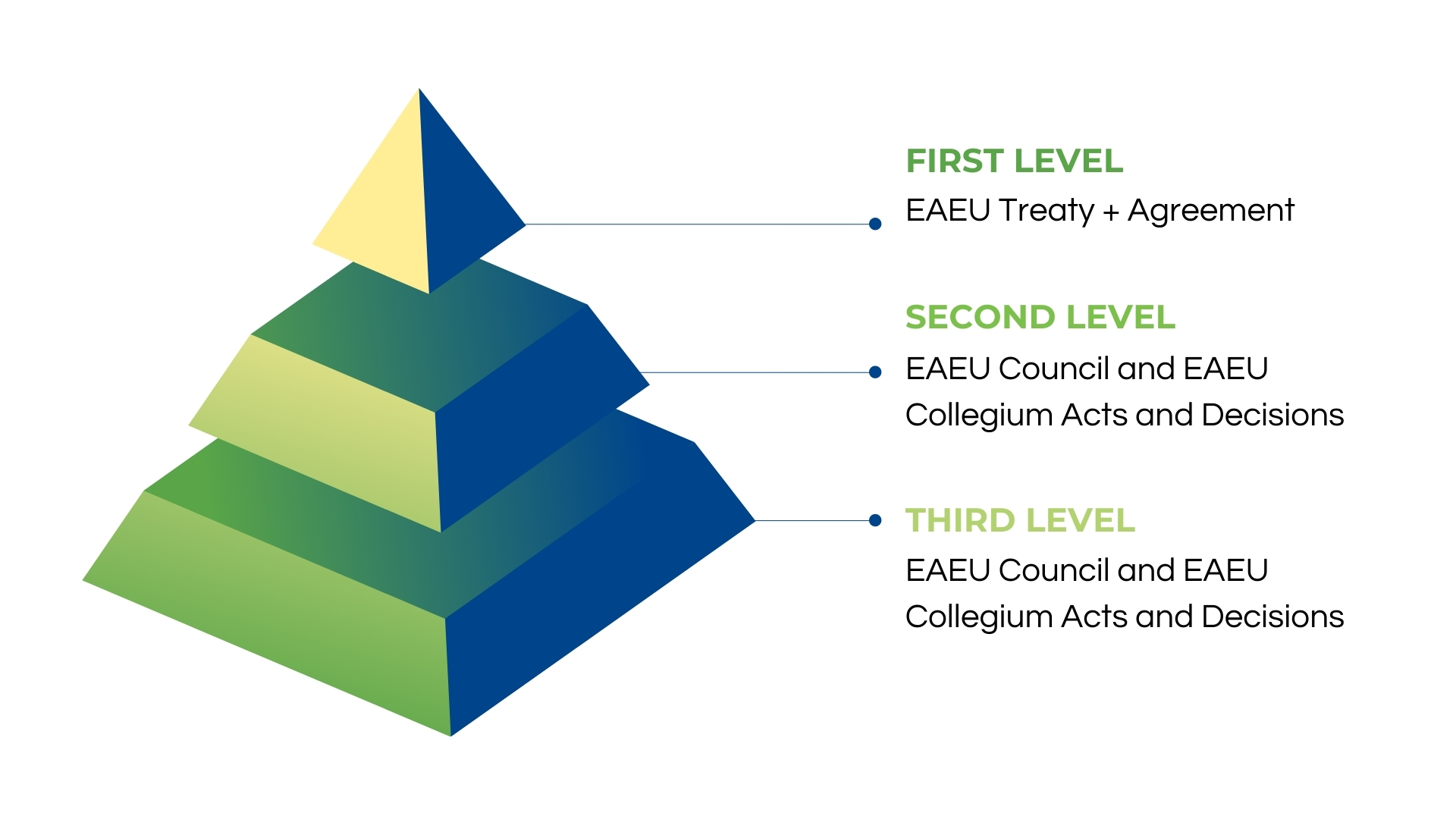
With Russian national registration, a medical device can circulate only in the Russian Federation, while the Authorized Representative of the Manufacturer is responsible only for the mentioned circulation. However, if a medical device is registered under EAEU legislation, it can circulate in all EAEU member states, while the Authorized Representative is responsible for all member states as well. In this case, the operation documents must be translated to all EAEU member states’ languages, not only Russian.
In addition, under Russian legislation, documents are approved by the Manufacturer, but under EAEU regulations, they can be approved by the Applicant instead. Nevertheless, the most crucial difference is number five, describing a new approach to quality, effectiveness and safety confirmation.
What Are the Changes in a Medical Device Registration Dossier?
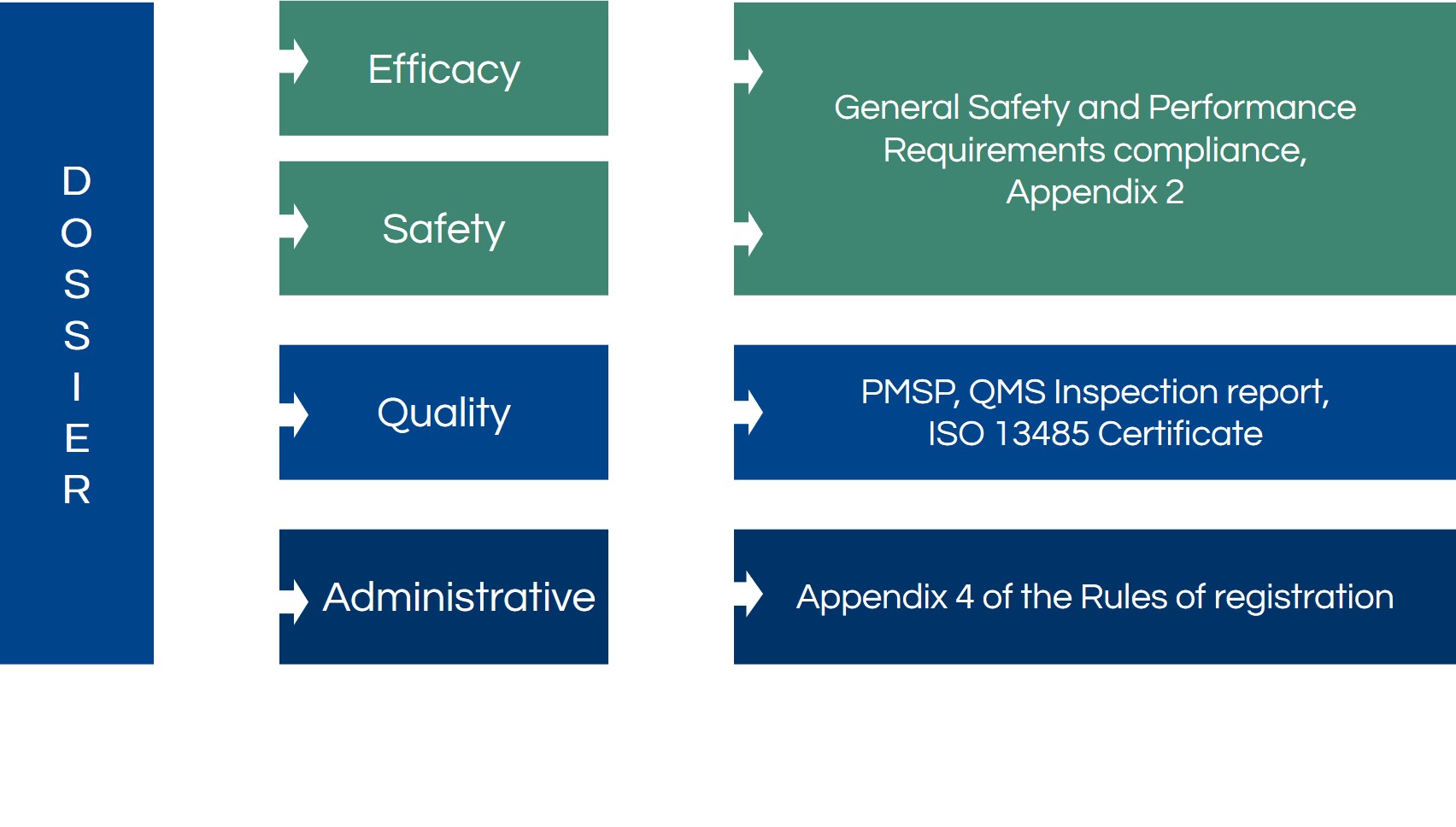
In current national Russian legislation, the medical device registration dossier contains the application document and approximately eleven additional documents, from technical testing reports to expert conclusion reports for stage 1 and 2. However, in EAEU legislation, a second application for the EAEU expertise and approximately thirty additional documents are necessary for the submission.
Furthermore, new types of documents are required, such as manufacturing plant inspection report, post-marketing surveillance plan, and expert conclusion of the reference country.
| NATIONAL RUSSIAN LEGISLATION |
EAEU LEGISLATION |
| 1. Application for the state registration and approx. 11 additional documents, including: |
1. Application for the EAEU registration
2. Application for the EAEU expertise, and approx. 30 additional documents, including:
|
| 2.Technical testing Report |
3. Technical testing Report |
| 3. Toxicology testing Report |
4. Toxicology testing Report |
| 4. Expert Conclusion I stage |
5. Clinical Trials Report |
| 5. Permission for Clinical evaluation |
6. Manufacturing Plant Inspection Report |
| 6. Clinical evaluation Report |
7. Post-marketing surveillance Plan |
| 7. Expert Conclusion II stage |
8. Expert Conclusion of Reference Country |

")






")

")
")